
ビラセプト錠250mgの添付文書

| 商品名: | ビラセプト錠250mg |
|---|---|
| 一般名: | ネルフィナビルメシル酸塩 |
| 略称 : | NFV |
添付文書の読み方
ここで提供している添付文書情報は、2009年11月12日現在の各医薬品の添付文書を基に作成したものです。書式等については、実際の添付文書と異なるところがあります。添付文書情報は随時更新されます。ご使用の際は、必ず最新の添付文書をご覧下さい。
また、記載されている内容には、専門的な情報が含まれています。文書内の、
この色の文字をクリックすると、別ウィンドウに読み方のアドバイスが表示されます。
この色の文字をクリックすると、別ウィンドウに重大な副作用の解説が表示されます。
この色の文字をクリックすると、別ウィンドウに副作用の症状とその類似語、定義の解説が表示されます。
記載されている情報をご覧になり、疑問などを持たれた場合は、医師・薬剤師にご相談ください。
このページを開いた時に目次が自動的に表示されなかったときは、ここをクリックしてください。
抗ウイルス化学療法剤
ビラセプト®錠250mg
Viracept®Tab.250mg
(ネルフィナビルメシル酸塩錠)
2009年9月改訂(第16版)
日本標準商品分類番号
87625
劇薬
処方せん医薬品注1)
注1)注意-医師等の処方せんにより使用すること
貯法:室温保存
(「取扱い上の注意」の項参照)
使用期限:3年
(外箱及びラベルに表示の使用期限を参照のこと)
| 承認番号 | 22100AMX01388000 |
|---|---|
| 薬価収載 | 2009年9月 |
| 販売開始 | 1998年3月 |
 Page Top
Page Top【禁忌(次の患者には投与しないこと)】
(1)本剤の成分に対して過敏症の既往歴がある患者
(2)テルフェナジン,シサプリド,トリアゾラム,ミダゾラム,アルプラゾラム,ピモジド,バッカク誘導体,アミオダロン及びキニジン硫酸塩水和物を投与中の患者(「相互作用」の項参照)
(3)リファンピシンを投与中の患者(「相互作用」の項参照)
(4)エレトリプタン臭化水素酸塩を投与中の患者(「相互作用」の項参照)
(5)エプレレノンを投与中の患者(「相互作用」の項参照)
【組成・性状】
1.組成
ビラセプト錠250mgは,1錠中にネルフィナビル250mg(ネルフィナビルメシル酸塩として292.25mg)を含有するうすい青色のフィルムコーティング錠である。添加物としてクロスポビドン,ケイ酸カルシウム,ステアリン酸マグネシウム,青色2号,ヒプロメロース,トリアセチンを含有する。
2.性状
| 外形 | サイズ | 識別コード | ||||
|---|---|---|---|---|---|---|
| 上 | 下 | 側面 | 長径mm | 短径mm | 厚さmm | |
 |
 |
 |
約19.1 | 約6.4 | 約6.1 | VIRACEPT 250mg |
【効能・効果】
HIV感染症
【用法・用量】
通常,成人にはネルフィナビルとして1回1,250mgを1日2回,または1回750mgを1日3回食後に経口投与する。なお,投与に際しては必ず他の抗HIV薬と併用すること。
〈用法・用量に関連する使用上の注意〉
(1)本剤の使用法を必要以上に変更又は中止すると副作用の発現やHIVの耐性化を促進するおそれがある。〔プロテアーゼ阻害剤に対するHIVの交差耐性については十分な検討がなされていないので,本剤の投与中止後に投与されるプロテアーゼ阻害剤の活性に対してどのように影響するかは不明である。〕
(2)ジダノシンは食間に投与されることとされているので,ジダノシンと本剤を併用する場合は,ジダノシンの投与と2時間以上の間隔を空けて投与すること。
【使用上の注意】
1.慎重投与(次の患者には慎重に投与すること)
(1)肝機能障害のある患者。〔代謝機能の低下により,高い血中濃度が持続するおそれがある。〕
(2)血友病患者及び著しい出血傾向を有する患者。〔血友病患者において,本剤投与による加療中に,脳内出血,縦隔内出血の発現が報告されており,また,関節内出血,皮下出血等の出血事象の増加が報告されている。〕
2.重要な基本的注意
(1)本剤の使用に際しては,患者又はそれに代わる適切な者に,次の事項についてよく説明し同意を得た後,使用すること。
1)本剤はHIV感染症の根治療法薬でないことから,日和見感染症を含むHIV感染症の進展に伴う疾病を発症し続ける可能性があるので,本剤投与開始後の身体状況の変化については,すべて担当医に報告すること。
2)本剤の長期投与による影響については,現在のところ不明であること。
3)本剤による治療が,性的接触又は血液汚染等による他者へのHIV感染の危険を減少させることは明らかではないこと。
4)本剤は必ず食後に服用すること。(空腹時に服用すると吸収が約50%減少する。)
5)本剤の抗ウイルス効果を最大にするために,医師への相談なしで,本剤の服用を変更したり,中止しないこと。
(2)最も頻度の高い副作用は下痢である。下痢の発現機序については本剤の腸管運動亢進作用が示唆されている。海外の二重盲検比較試験において下痢を発現した463症例のうち,195例(42%)にロペラミドが投与されており,また,国内の臨床試験において,ロペラミドやタンニン酸製剤などの投与により,下痢が改善若しくは消失したとの報告がある。なお,下痢発現例と非発現例において,血中濃度に有意な差は認められておらず,本剤の有効性には影響は認められていない。
(3)国内での臨床試験(38例)において,発疹が6例(15.8%)及び斑丘疹が3例(7.9%)発現している。これらの発現日は本剤の投与を開始してから,平均10日後(7~13日後及び9~10日後)である。本剤に起因すると考えられる発疹及び斑丘疹が発現した場合には,本剤の投与を中止し,他の適切な療法を行うこと。なお,やむを得ず本剤の投与を再開する場合には,発疹,斑丘疹が軽快したことを確認のうえ,慎重に投与すること。
(4)本剤を含む抗HIV薬の多剤併用療法を行った患者で,免疫再構築症候群が報告されている。投与開始後,免疫機能が回復し,症候性のみならず無症候性日和見感染(マイコバクテリウムアビウムコンプレックス,サイトメガロウイルス,ニューモシスチス等によるもの)等に対する炎症反応が発現することがあるので,これらの炎症性の症状を評価し,必要時には適切な治療を考慮すること。
(5)抗HIV薬の使用により,体脂肪の再分布/蓄積があらわれることがあるので,異常が認められた場合には適切な処置を行うこと。
3.相互作用
本剤は,主として肝代謝酵素CYP3A4及び一部CYP2C19で代謝され,また,CYP3A4の阻害作用を持つ。
(1)併用禁忌(併用しないこと)
| 薬剤名等 | 臨床症状・措置方法・機序・危険因子 |
|---|---|
|
本剤のチトクロームP450(CYP3A4)に対する競合により,これら薬剤の代謝が抑制され,重篤な又は生命に危険を及ぼすような事象(QT延長,Torsade de Pointes等の不整脈や持続的な鎮静)が起こる可能性がある。 |
|
本剤の血中濃度が20~30%に低下する。リファンピシンの投与を受けた患者に本剤を投与する場合には,少なくとも2週間の間隔をおくことが望ましい。 |
|
エレトリプタンの血中濃度が上昇する可能性がある。 |
|
エプレレノンの血中濃度が上昇する可能性がある。 |
(2)併用注意(併用に注意すること)
| 薬剤名等 | 臨床症状・措置方法・機序・危険因子 |
|---|---|
| インジナビル サキナビル |
本剤及びインジナビル又はサキナビルの血中濃度が上昇する。両剤の併用による安全性及び有効性は確立していない。 |
| リトナビル | 本剤の血中濃度が上昇する。両剤の併用投与による安全性及び有効性は確立していない。 |
| アンプレナビル | 本剤の血中濃度が上昇し,アンプレナビルの血中濃度が変動する。両剤の併用による安全性及び有効性は確立していない。 |
| デラビルジン | 本剤の血中濃度が約2倍に上昇し,デラビルジンの血中濃度が約30%低下する。 |
| リファブチン | 本剤の血中濃度が低下し,リファブチンの血中濃度が上昇するため,リファブチンの投与量を半量以下に減量する。 |
| エチニルエストラジオール又はノルエチステロンを含む経口避妊薬 | エチニルエストラジオールとノルエチステロンの血中濃度が低下するため,本剤投与中は他の避妊法の追加又は変更を行う。 |
| フェノバルビタール フェニトイン カルバマゼピン |
本剤の血中濃度を低下させるおそれがあり,これら薬剤の血中濃度が変動する可能性がある。 |
| シルデナフィル | シルデナフィルの血中濃度が上昇する可能性がある。 |
| シンバスタチン | シンバスタチンのAUCが約6倍に上昇するとの報告があり,横紋筋融解症,ミオパシー等の副作用が発現するおそれがあることから,本剤とシンバスタチンとの併用は避けることが望ましい。 |
| アトルバスタチン | アトルバスタチンのAUCが約1.7倍に上昇するとの報告がある。 |
| タクロリムス シクロスポリン エベロリムス |
これら薬剤の血中濃度が上昇する可能性がある。 |
| セイヨウオトギリソウ(St.John's Wort,セント・ジョーンズ・ワート)含有食品 | 本剤の代謝が促進され血中濃度が低下するおそれがあるので,本剤投与時はセイヨウオトギリソウ含有食品を摂取しないよう注意すること。 |
| アジスロマイシン | アジスロマイシンの血中濃度が約2倍に上昇するとの報告がある。 |
| ボリコナゾール | 本剤及びボリコナゾールの血中濃度が上昇するおそれがある。 |
| オメプラゾール | 本剤の血中濃度が低下するおそれがある。 |
| フルチカゾン | フルチカゾンの血中濃度が上昇する可能性がある。 |
| トラゾドン | トラゾドンの血中濃度が上昇する可能性がある。 |
4.副作用
海外での臨床試験において,1,177例中965例(82%)に副作用が認められ,主なものは,下痢,嘔気,腹部膨満感,後天性リポジストロフィー,頭痛,脱力感,腹痛,発疹等であった。
また,国内での臨床試験において,総症例38例中20例(53%)に副作用が認められ,主なものは,下痢,嘔気,発疹,そう痒感,腹痛であった。
(承認時及び用法・用量に係る一変承認時の集計)
(1)重大な副作用
- 1)糖尿病,血糖値の上昇(頻度不明注2))
- 本剤の投与により,糖尿病,糖尿病の悪化及び血糖値の上昇が報告されており,その中には重篤な症例やケトアシドーシスを伴う症例も報告されているので,このような症状があらわれた場合には,投与を中止するなど適切な処置を行うこと。
- 2)出血傾向(頻度不明注2))
- 血友病患者において,本剤の投与による加療中に, 脳内出血,縦隔内出血の発現が報告されており,また,関節内出血,皮下出血等の出血事象の増加が報告されているので,このような症状があらわれた場合には適切な処置を行うこと。また必要に応じて,血液凝固因子の投与などの処置を行うこと。
(2)その他の副作用
次のような症状があらわれた場合には,症状に応じて適切な処置を行うこと。(下表の頻度は海外及び国内の臨床試験より算出した。)
| 種類\頻度 | 2%以上 | 2%未満 | 頻度不明注2) |
|---|---|---|---|
| 全身 | 後天性リポジストロフィー(体脂肪の再分布/蓄積;胸部,体幹部の脂肪増加,末梢部の脂肪減少,野牛肩)(9.3%),頭痛(8.1%),脱力感(7.2%) | 悪液質,疼痛,体重減少,体重増加,けん怠感,発熱,背部痛,胸部痛,悪寒,疲労感 | |
| 循環器 | 血管拡張,浮腫,頻脈,末梢性浮腫,動悸 | ||
| 消化器 | 下痢(66.8%),嘔気(13.3%),腹部膨満感(10.9%),腹痛(6.8%),嘔吐(4.0%),鼓腸(3.7%),消化不良(2.5%),食欲不振(2.4%) | おくび,胃炎,嚥下障害,便秘,口渇,口内炎,直腸の異常,食欲亢進,便異常,舌の異常,排便障害,口腔内違和感 | 膵炎 |
| 血液,リンパ系 | 白血球減少,リンパ節腫脹,好中球減少,貧血 | ||
| 代謝,栄養系 | 高脂血症,高尿酸血症,高コレステロール血症 | 高トリグリセリド血症 | |
| 肝臓 | ALT(GPT)上昇,AST(GOT)上昇,CK(CPK)上昇,γ-GTP上昇,LDH上昇,総ビリルビン上昇,Al-p上昇,肝機能障害 | ||
| 筋骨格系 | 筋肉痛,関節痛,下肢の痙攣 | ||
| 精神神経系 | 眩暈(2.9%) | 抑うつ,傾眠,不眠,情緒不安,不安,異常思考,睡眠異常,健忘症,混乱,多動 | |
| 呼吸器 | 咽頭炎,呼吸困難 | ||
| 皮膚 | 発疹(4.9%),そう痒感(2.3%) | 斑丘疹,発汗,皮膚乾燥,皮膚の異常,毛包炎,ざ瘡,蕁麻疹 | 多形紅斑 |
| 感覚器 | 感覚異常(3.5%) | 味覚異常,視覚異常,眼の異常,嗅覚異常,味覚喪失 | |
| 泌尿器 | 尿の異常,頻尿,血尿,排尿障害 | ||
| 生殖器 | 月経異常,インポテンス |
注2)市販後調査,自発報告等にて報告された副作用
5.高齢者への投与
高齢者における安全性及び有効性は確立していない。一般に高齢者では生理機能が低下しているので,注意すること。
6.妊婦,産婦,授乳婦等への投与
(1)妊婦又は妊娠している可能性のある婦人には,治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。〔妊娠中の投与に関する安全性は確立していない。〕
(2)本剤服用中は授乳を中止させること。〔動物実験(ラット)で乳汁中への移行が報告されている。〕
7.小児等への投与
低出生体重児,新生児,乳児,幼児又は小児に対する安全性は確立していない(使用経験が少ない)。
8.過量投与
本剤の過剰量をごく短時間に服用した症例の報告は殆どない。本剤の特別な解毒法はない。過剰に投与した場合,吸収されていない薬剤は嘔吐,胃洗浄又は活性炭で除去する。本剤はタンパク結合率が高いため,血中からの除去法として透析は不適切である。
9.その他の注意
(1)ラットを用いた癌原性試験(2年間)において,甲状腺ろ胞上皮の増殖性病変(過形成,腺腫,腺癌)が,300mg/kg投与の雄及び1,000mg/kg投与の雌雄で発現したとの報告がある。
(2)本剤投与中に,本剤の添加物に由来する青色の残渣が,便中に観察されることがある。
【薬物動態】
〈日本人における成績〉
1.血中濃度・排泄1),2)
健康成人男子に本剤250,500,750,1,000mg(各々6名)を食後単回投与した場合,未変化体の血漿中濃度は投与約3~5時間後に最高濃度に達し,消失半減期は約2~5時間であった。(下表)
| 投与量注3)(mg) | 例数 | Tmax(h) | Cmax(μg/mL) | T1/2(h) | AUC(μg・h/mL) |
|---|---|---|---|---|---|
| 250 | 6 | 3.3±0.8 | 0.59±0.15 | 2.1±0.1 | 3.10±1.17 |
| 500 | 6 | 3.5±0.5 | 1.91±0.64 | 3.4±0.9 | 16.30±6.44 |
| 750 | 6 | 4.7±1.0 | 4.86±0.71 | 4.8±0.9 | 47.91±10.75 |
| 1,000 | 6 | 4.5±1.2 | 6.66±2.33 | 4.1±0.8 | 65.53±24.82 |
平均値±S.D.
注3)本剤の承認された1回用量は1250mg又は750mgである
また,投与後24時間までの未変化体の平均尿中排泄率はいずれの投与量においても0.2%以下であった。
一方,健康成人男子(6名)に本剤750mgを1日3回7日間食後経口投与した場合,未変化体のトラフ値(毎朝第1回目投与直前の血漿中濃度)は初日投与後から4日目まで減少したが,それ以降はほぼ定常状態となった。なお,定常状態における平均のトラフ値は約3μg/mLであった。
また,未変化体の累積尿中排泄率は単回投与時と同様に微量であり,投与後48時間までに総投与量の0.26%が排泄された。
2.食事の影響1)
健康成人男子に本剤500mg注3)を空腹時単回投与した場合のCmax及びAUCは,食後単回投与時と比べそれぞれ51%,41%に減少した。
また,未変化体の平均尿中排泄量も食後投与時の約1/2に減少した。
注3)本剤の承認された1回用量は1250mg又は750mgである
〈外国人における成績(参考)〉
1.血中濃度
HIV陽性患者を対象に,本剤1,250mg 1日2回又は750mg 1日3回を反復投与した場合の,投与28日目の薬物動態パラメータを下表に示した。
| 用法・用量 | 例数 | AUC24注4) (μg・h/mL) |
Cmax (μg/mL) |
Ctrough1注5) (μg/mL) |
Ctrough2注6) (μg/mL) |
|---|---|---|---|---|---|
| 1,250mg 1日2回投与 |
10 | 52.8±15.7 | 4.0±0.8 | 2.2±1.3 | 0.7±0.4 |
| 750mg 1日3回投与 |
11 | 43.6±17.8 | 3.0±1.6 | 1.4±0.6 | 1.0±0.5 |
注4)定常状態における24時間AUC値
注5)朝の投薬前のトラフ値
注6)朝の投薬12時間後(1,250mg 1日2回投与)又は8時間後(750mg 1日3回投与)のトラフ値
2.食事の影響
健康成人に本剤1,250mgを投与した場合の熱量及び脂肪含有率における薬物動態パラメータを下表に示した。
| 熱量(kcal) | 脂肪含有率(%) | 例数 | AUC0-∞(μg・h/mL) | Cmax(μg/mL) | Tmax(h) |
|---|---|---|---|---|---|
| 0 | 0 | 22 | 10.6 | 1.57 | 2.18 |
| 125 | 20 | 21 | 23.1 | 3.16 | 3.02 |
| 500 | 20 | 22 | 33.4 | 3.67 | 3.87 |
| 1,000 | 50 | 23 | 55.3 | 5.20 | 3.98 |
また,健康成人に本剤1,250mgを投与した場合の食事の脂肪含有率における薬物動態パラメータを下表に示した。
| 熱量(kcal) | 脂肪含有率(%) | 例数 | AUC0-∞(μg・h/mL) | Cmax(μg/mL) | Tmax(h) |
|---|---|---|---|---|---|
| 0 | 0 | 22 | 10.7 | 1.63 | 2.42 |
| 500 | 20 | 22 | 32.7 | 4.04 | 4.19 |
| 500 | 50 | 22 | 54.6 | 6.16 | 4.48 |
3.タンパク結合率・血球移行率(in vitro)
本剤はin vitro試験において,1.0~21.5μg/mLの濃度範囲で98.7~99.3%がヒト血清タンパクと結合し,そのタンパクは主にアルブミン,α1-酸性糖タンパクであった。また,本剤の血球移行率は4.3μg/mLの添加濃度で12.5%であった。
4.代謝3)
健康成人男子に14C標識ネルフィナビルメシル酸塩を単回経口投与し,糞中及び血漿中の代謝物を検索した結果,糞中では,ネルフィナビルは主にt-ブチル基の水酸化,デカヒドロイソキノリン環上の水酸化,ベンゾイル環上の水酸化(カテコールの生成),カテコールのメチル化等の代謝物が検出された。血漿中の主要代謝物はt-ブチル基の水酸化物であり,抗ウイルス活性(in vitro)は未変化体と同程度であった。
In vitroの試験において,本剤は肝ミクロソームにより酸化的な代謝反応を受け,その代謝はチトクロームP450(CYP)3A4の阻害剤で最も大きく阻害された。
5.排泄
健康成人男子(4名)に750mgの14C標識ネルフィナビルメシル酸塩を単回経口投与した場合,投与後120時間までに投与した放射能の78.2%が糞便中に,1.6%が尿中にそれぞれ排泄された。
6.薬物相互作用4)
ヒトチトクロームP450アイソザイム(CYP3A4,CYP2C19,CYP2D6,CYP2C9,CYP1A2及びCYP2E1)に対するネルフィナビルの阻害活性をin vitro試験により検討した結果,ネルフィナビルが治療的薬効域において,CYP3A4を阻害した。
抗HIV剤を含む主な薬剤との併用による血中濃度(AUC,Cmax)への影響を以下に示す。
| 併用薬名 | ネルフィナビル投与量 | 例数 | 併用薬血中濃度の変化量 | |
|---|---|---|---|---|
| AUC | Cmax | |||
| ラミブジン 150mg単回投与 |
750mg 8時間毎1日3回投与×7-10日 | 11 | ↑9% | ↑34% |
| サニルブジン 30-40mg1日2回投与×56日 |
750mg 1日3回投与×56日 | 8 | 変化なし | 変化なし |
| ジドブジン 200mg単回投与 |
750mg 8時間毎1日3回投与×7-10日 | 11 | ↓34% | ↓31% |
| インジナビル 800mg単回投与 |
750mg 8時間毎1日3回投与×7日 | 6 | ↑43% | 変化なし |
| リトナビル 500mg単回投与 |
750mg 8時間毎5回 | 10 | 変化なし | 変化なし |
| サキナビル(軟カプセル) 1,200mg単回投与 |
750mg 1日3回投与×4日 | 14 | ↑416% | ↑179% |
| エチニルエストラジオール 35μg1日1回投与×15日 |
750mg 8時間毎1日3回投与×7日 | 12 | ↓49% | ↓30% |
| ノルエチステロン 0.4mg1日1回投与×15日 |
750mg 8時間毎1日3回投与×7日 | 12 | ↓20% | 変化なし |
| リファブチン 150mg1日1回投与×8日 |
750mg 8時間毎1日3回投与×7-8日 | 12 | ↑83%注7) | ↑19%注7) |
| リファブチン 300mg1日1回投与×8日 |
750mg 8時間毎1日3回投与×7-8日 | 10 | ↑200% | ↑145% |
| シンバスタチン 20mg1日1回投与×28日 |
1,250mg 12時間毎1日2回投与×14日 | 16 | ↑504% | ↑517% |
| アトルバスタチン 10mg1日1回投与×28日 |
1,250mg 12時間毎1日2回投与×14日 | 15 | ↑74% | ↑122% |
| アジスロマイシン 1,200mg単回投与 |
750mg 8時間毎1日3回投与×11日 | 12 | ↑108% | ↑107% |
↑増加 ↓減少
注7)リファブチン300mg 1日1回投与×7日 単独投与に対する変化量
| 併用薬名 | ネルフィナビル投与量 | 例数 | ネルフィナビル血中濃度の変化量 | |
|---|---|---|---|---|
| AUC | Cmax | |||
| ジダノシン 200mg単回投与 |
750mg 単回投与 | 10 | 変化なし | 変化なし |
| ジドブジン200mg単回投与 +ラミブジン150mg単回投与 |
750mg 8時間毎1日3回投与×7-10日 | 11 | 変化なし | 変化なし |
| インジナビル 800mg8時間毎1日3回投与×7日 |
750mg 単回投与 | 6 | ↑79% | ↑30% |
| リトナビル 500mg12時間毎3回 |
750mg 単回投与 | 10 | ↑156% | ↑44% |
| サキナビル(軟カプセル) 1,200mg1日3回投与×4日 |
750mg 単回投与 | 14 | ↑18% | 変化なし |
| ケトコナゾール 400mg1日1回投与×7日 |
500mg注8) 8時間毎1日3回投与×5-6日 | 12 | ↑33% | ↑23% |
| リファブチン 150mg1日1回投与×8日 |
750mg 8時間毎1日3回投与×7-8日 | 11 | ↓23% | ↓18% |
| リファブチン 150mg1日1回投与×8日 |
1,250mg 12時間毎1日2回投与×7-8日 | 11 | 変化なし | 変化なし |
| リファブチン 300mg1日1回投与×8日 |
750mg 8時間毎1日3回投与×7-8日 | 10 | ↓32% | ↓24% |
| リファンピシン 600mg1日1回投与×7日 |
750mg 8時間毎1日3回投与×5-6日 | 12 | ↓82% | ↓74% |
| アジスロマイシン 1,200mg単回投与 |
750mg 8時間毎1日3回投与×11日 | 12 | ↓16% | ↓10% |
| オメプラゾール 40mg1日1回投与×4日 |
1,250mg 1日2回投与×4日 | 19 | ↓36% | ↓37% |
↑増加 ↓減少
注8)本剤の承認された1回用量は1250mg又は750mgである
【臨床成績】
〈日本人における成績〉5)
日本人のHIV感染症患者38例に対して,ネルフィナビルとして1回750mgを1日3回,ジドブジンを含む逆転写酵素阻害剤と24週間併用投与を行った。その結果,免疫学的評価の主要項目であるCD4リンパ球数は,投与開始後24週目において基準値より75cells/mm3の上昇を示した。また,ウイルス学的評価の主要項目である血漿中HIV RNA量は,検出限界(100copies/mL)以下となった症例のHIV RNA量を50copies/mLと仮定したとき,投与開始後2週目以降基準値からの持続的な低下を示し,投与開始後4週目において平均-1.41log10copies/mLと最大の低下を示した。また,投与開始後12週目及び24週目においてそれぞれ平均-1.11log10copies/mL及び-1.26log10copies/mLの低下を示し,これらの値にはそれぞれ統計学的な有意差(p<0.0001及びp=0.0004)が認められた。血漿中HIV RNA量が検出限界以下となった症例の割合は,投与開始後12週目で19.4%,24週目で28.6%を示した。
〈外国人における成績〉(参考)
1.511試験
抗レトロウイルス剤による治療経験のない297例のHIV感染症患者を対象に,ジドブジン(200mg,1日3回投与)及びラミブジン(150mg,1日2回投与)を基礎治療薬として本剤の500mg注9)(500mg群),750mg(750mg群)及びプラセボ(コントロール群)を1日3回48週間投与する二重盲検比較試験により検討した。
患者の年齢の中間値は35歳(21~63歳),78%が白人,89%が男性であり,開始時の平均CD4リンパ球数は288cells/mm3,平均血漿中HIV RNA量は5.21log10copies/mLであった。投与後48週目のCD4リンパ球数の平均増加量は,198cells/mm3(750mg群),192cells/mm3(500mg群),127cells/mm3(コントロール群)であった。
また,投与48週後までの血漿中HIV RNA量が400copies/mL未満であった患者の比率注10)の推移を図に示した。
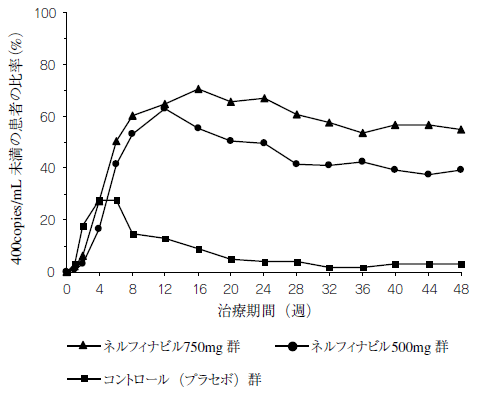
注9)本剤の承認された1回用量は1250mg又は750mgである
注10)理由にかかわらず試験を中止した患者及び効果不十分の理由で薬剤を変更した患者については,それ以降の期間の血漿中HIV RNA量を400copies/mL以上とみなした。
2.542試験
核酸系逆転写酵素阻害剤の使用経験が6カ月以下で,かつHIVプロテアーゼ阻害剤の使用経験のないHIV感染症患者を対象に,サニルブジン(30~40mg,1日2回投与)及びラミブジン(150mg,1日2回投与)を基礎治療薬として本剤750mgを1日3回(TID群:210例),又は1,250mgを1日2回(BID群:344例)投与する無作為化オープン試験を実施した。
患者の平均年齢はTID群で37歳,BID群で36歳,TID群の91%が白人で82%が男性,BID群の91%が白人で85%が男性であった。開始時の平均CD4リンパ球数はTID群,BID群で各々304cells/mm3,292cells/mm3,平均血漿中HIV RNA量はTID群,BID群で各々5.1log10copies/mL,5.0log10copies/mLであった。投与後48週目のCD4リンパ球数の平均増加量は,192cells/mm3(TID群),212cells/mm3(BID群)であり有意差はなかった。
また,投与48週後までの血漿中HIV RNA量が400copies/mL未満であった患者の比率注11)の推移を図に示した。
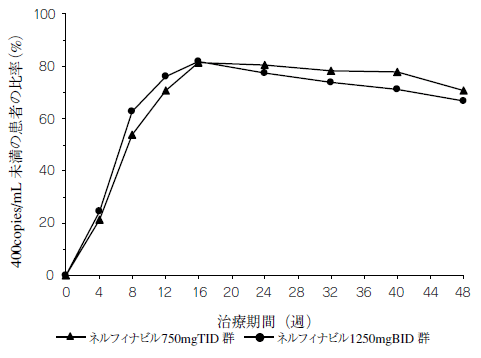
注11)理由にかかわらず試験を中止した患者及び効果不十分の理由で薬剤を変更した患者については,それ以降の期間の血漿中HIV RNA量を400copies/mL以上とみなした。
3.薬物血中濃度と有効性・副作用発現の関連について
本剤を単剤で投与した503試験の65例において,各症例の朝の投薬前の平均トラフ値と抗HIV活性を比較検討した結果,投与開始後4週間に血漿中HIV RNA量が2.0log10copies/mL以上低下した症例,1.0~2.0log10copies/mL低下した症例,一過性に1.0log10copies/mL以上低下したが4週以内に再上昇した症例及び低下が1.0log10copies/mL未満であった症例の4群に分けた場合の平均未変化体トラフ値は,各々2.10±1.10μg/mL(n=25),1.86±0.74μg/mL(n=27),1.37±0.95μg/mL(n=9)及び0.93±0.30μg/mL(n=4)であり,本剤の有効性は血中濃度依存的であることが推察された。また,ジドブジン及びラミブジンとの併用試験(511試験)においても,本剤の有効性が血中濃度依存的であることが認められている。
一方,同じ503試験において,本剤の主たる副作用である下痢,皮疹及び嘔気・嘔吐が発現した症例と発現しなかった症例における朝の投薬前の平均未変化体トラフ値を比較検討した結果,下痢発現例及び非発現例で各々1.86±0.96μg/mL(n=43)及び1.75±0.97μg/mL(n=22),皮疹発現例及び非発現例で各々1.81μg/mL(n=2)及び1.83±0.97μg/mL(n=63),嘔気・嘔吐発現例及び非発現例で各々2.08±1.20μg/mL(n=4)及び1.81±0.94μg/mL(n=61)といずれも平均未変化体トラフ値に有意差は認められず,有害事象の発現は本剤のトラフ値に依存していないと推察された。
【薬効薬理】
1.作用機序
本剤は,HIV-1由来のプロテアーゼの活性を選択的に阻害する。本剤は,ヒト由来のアスパラギン酸プロテアーゼ(レニン,ペプシン,ガストリン,カテプシン等)に対する阻害活性はほとんどなく,HIVプロテアーゼに対して高い酵素特異性を示す。本剤は,プロテアーゼの活性中心において,HIV前駆体ポリタンパク質と競合してプロテアーゼ活性を阻害し,その結果,ウイルス粒子の成熟過程において,HIV前駆体ポリタンパク質の切断が妨げられ,感染性を持つHIVの産生を抑制する。
2.抗ウイルス作用(in vitro)6),7)
HIV-1(RF及びⅢB)株とヒトTリンパ球系細胞(CEM-SS及びMT-2)株による急性感染系において,本剤は31及び43nmol/Lの濃度で,ウイルス増殖を50%阻害(無処置ウイルス感染の対照と比較)した。HIV-2(ROD)株とヒトTリンパ球系細胞株の系においても,本剤はウイルス増殖を9nmol/Lで50%阻害した。単球指向性ウイルス株(Ba-L)を用いた試験では,培養ヒト単球/マクロファージにおいても,同様に本剤によるHIV-1感染の阻害が認められた。更に,ジドブジンあるいは非ヌクレオシド系逆転写酵素阻害剤に対する耐性HIVを含むHIV臨床分離株で感染させたヒトTリンパ球系細胞(MT-2)株の系において,本剤は30~60nmol/Lでウイルス増殖を50%阻害した。また,ヒトTリンパ球系細胞株とHIV-1(RF)株の感染系において,本剤は逆転写酵素阻害剤(ジドブジン,ラミブジン等)との併用により,相乗的あるいは相加的なHIV増殖抑制作用を示した。
3.薬剤耐性8)
本剤が投与された患者において,本剤に対する感受性が低下したHIVが単離された。本剤に対する耐性HIVの発現は,HIVプロテアーゼのアミノ酸置換に基づくことが確認されており,プロテアーゼ領域の30番目のアミノ酸の変異が最も頻度が高く,本剤に対する耐性化に最も重要であることが判っている。また,その他さらに数ヵ所に変異がおこる場合があることも確認されている。なお,30番目のアミノ酸に変異を有する耐性ウイルスの発現頻度は,逆転写酵素阻害剤であるジドブジンとラミブジンを併用した場合に著しく抑制されることが認められている。
4.交差耐性8)
本剤に対する感受性が低下した30番目のアミノ酸変異をもつ耐性ウイルスは,他のプロテアーゼ阻害剤に対する感受性を維持していることが確認されている。また,他のプロテアーゼ阻害剤に対する耐性を発現した患者から分離したウイルスのうち,その61%(14/23株)は本剤に対する感受性を維持していることが確認されているが,遺伝子変異との関係については十分な検討はなされていない。本剤とHIV逆転写酵素阻害剤との間の交差耐性については,薬剤の作用点が異なること,及びジドブジン抵抗性HIV株やピリジノン系の非核酸系逆転写酵素阻害剤抵抗性HIV株に対して本剤の抗ウイルス作用の減弱が認められないことから,発現する可能性は低いと推定されている。
5.その他(参考)
モルモット全身性アナフィラキシー反応(感作sc,誘発iv)において弱い陽性反応が認められたが,臨床投与経路である経口投与下におけるモルモット全身性アナフィラキシー反応は,陰性であった。(感作po,誘発iv)
また,モルモットPCA反応及びマウスPCA反応はいずれも陰性であった。
【有効成分に関する理化学的知見】
- 一般名
- ネルフィナビルメシル酸塩 Nelfinavir mesilate
- 化学名
- (-)-(3S, 4aS, 8aS)-N-tert-butyl-2-[(2R, 3R)-2-hydroxy-3-(3-hydroxy-2-methylbenzoylamino)-4-(phenylthio)butyl]decahydroisoquinoline-3-carboxamide monomethanesulfonate
- 分子式
- C32H45N3O4S・CH4O3S
- 分子量
- 663.89
- 化学構造式
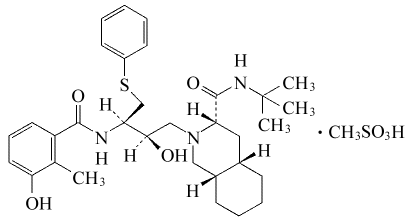
- 性状
- 白色~帯黄白色の粉末で,わずかに刺激性のにおいがある。
本品はメタノール,エタノール(99.5),アセトニトリルに溶けやすく,クロロホルムにやや溶けにくく,水(脱イオン水)に極めて溶けにくく,ジエチルエーテルにほとんど溶けない。 - 融点
- 融点を示さず,100~200℃で分解する。
【取扱い上の注意】
本剤は吸湿しやすいので,開栓後は,湿気を避けて保存すること。
【承認条件】
- 1.本剤を使用する場合は,皮疹に留意して,皮疹等の副作用が発生した場合には必ず副作用報告を行うよう,また,治療にあたっては,本剤は市販後調査において薬剤に関する科学的なデータを収集することとされていること等について患者に十分な説明を行い,インフォームド・コンセントを得るよう医師に対して要請すること。
- 2.本剤の皮疹及び下痢等の副作用については,その発生機序及び対処方法について調査,検討を継続し,検討結果を定期的(1年に1回程度を目途)に報告すること。
-
3.今後,再審査期間の終了までは,国内で使用される症例に関しては,可能な限り全投与症例を市販後調査の対象とし,薬物代謝酵素等の患者背景,臨床効果,副作用,薬物相互作用等に関してデータの収集を行い,再審査の申請資料として提出すること。
なお,データの収集においては,特に,本剤の副作用の発生機序及び対処方法の検討に資するデータに留意すること。 - 4.市販後,本薬の使用実態について詳細に調査を行い,他剤との併用における本剤の安全性,有効性に関する情報収集を実施し,定期的に報告すること。
- 5.実施予定及び実施中の臨床試験については,可及的速やかに成績及び解析結果を提出すること。
【包装】
ビラセプト®錠250mg:300錠/瓶
【主要文献】
- 木村哲, 他: 臨床医薬 14(11)1989, 1998
- 木村哲, 他: 臨床医薬 14(11)2005, 1998
- Zhang K. et al.: Antimicrob. Agents Chemother. 45(4)1086, 2001
- Lillibridge J. H. et al.: Drug Metab. Dispos. 26(7)609, 1998
- 木村哲, 他: 医学のあゆみ 192(9)915, 2000
- Patick A. K. et al.: Antimicrob. Agents Chemother. 40(2)292, 1996
- Patick A. K. et al.: Antimicrob. Agents Chemother. 41(10)2159, 1997
- Patick A. K. et al.: Antimicrob. Agents Chemother. 42(10)2637, 1998
【文献請求先】
中外製薬株式会社 医薬情報センター
〒103-8324 東京都中央区日本橋室町2-1-1
TEL 0120-189706
FAX 0120-189705
http://www.chugai-pharm.co.jp
日本たばこ産業株式会社 医薬事業部 医薬情報部
〒105-8422 東京都港区虎ノ門二丁目2番1号
- 製造販売元
- 日本たばこ産業株式会社
- 東京都港区虎ノ門二丁目2番1号
- 販売
- 中外製薬株式会社
- 東京都中央区日本橋室町2-1-1
| おくすりガイド|
おくすりガイド|